Figures were modified from Macosko et al., Cell, 2015 for educational purposes.
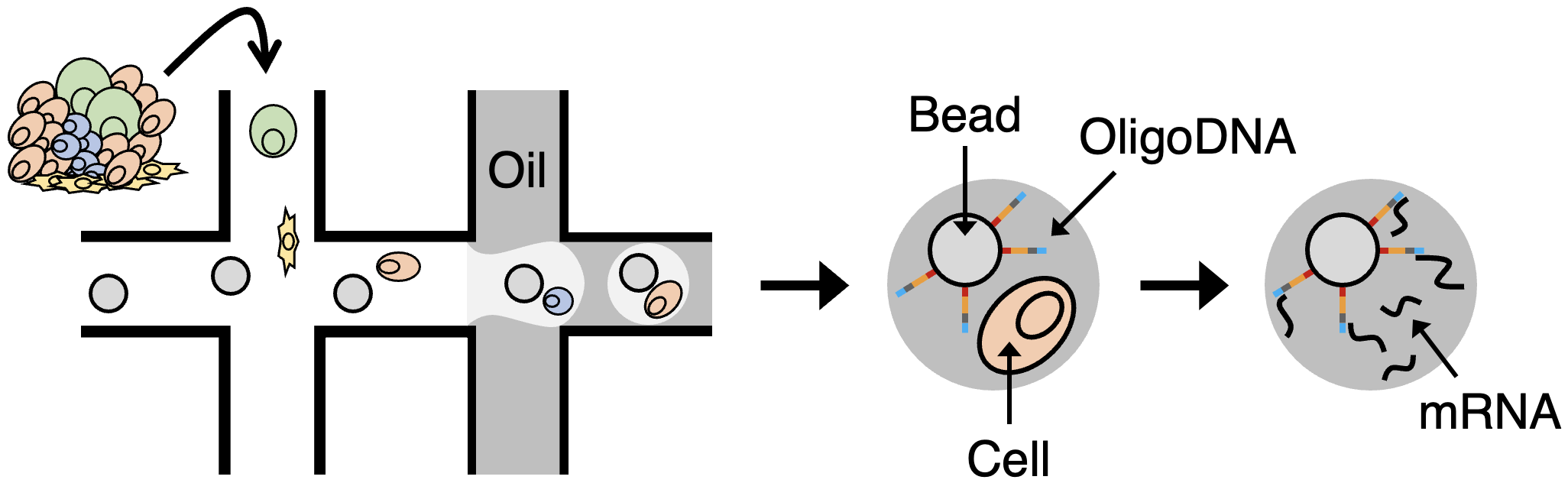
Step 1: Cell and Bead Encapsulation
Single cells and barcoded microparticles (beads) are encapsulated into aqueous droplets. Each droplet contains one cell and one bead, along with a lysis buffer that immediately lyses the cell and releases RNA.
Step 2: RNA Capture via Bead-bound polyT
Each bead is coated with many oligoDNA primers containing a polyT sequence, which hybridizes to the polyA tail of RNA molecules. Although the term "mRNA" is often used, nascent (unspliced) RNA with polyA tails may also be captured, which can be used for estimating transcription dynamics (velocyto).
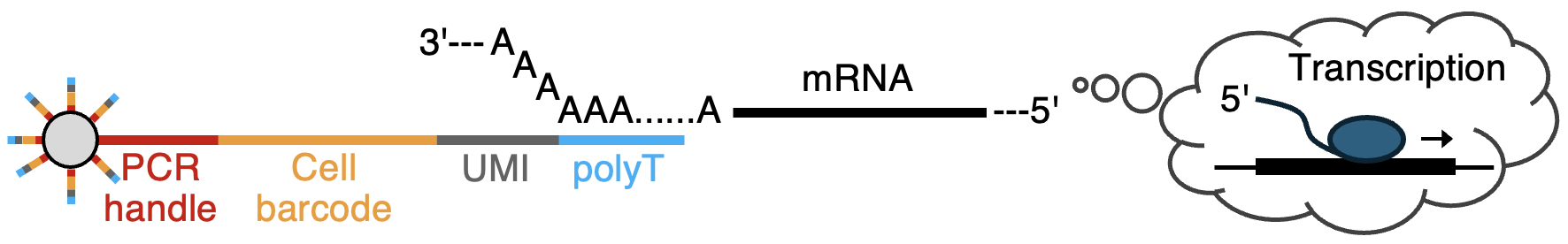
Each oligoDNA primer on the bead contains the following structure:
5' - PCR handle - Cell Barcode (12 bp) - UMI (8 bp) - polyT (T27) - 3'
where
Step 3: Reverse Transcription (RT)
The captured RNA is reverse transcribed into cDNA using the bead-bound oligo as a primer. This generates cDNA that is partially hybridized with the original primer.
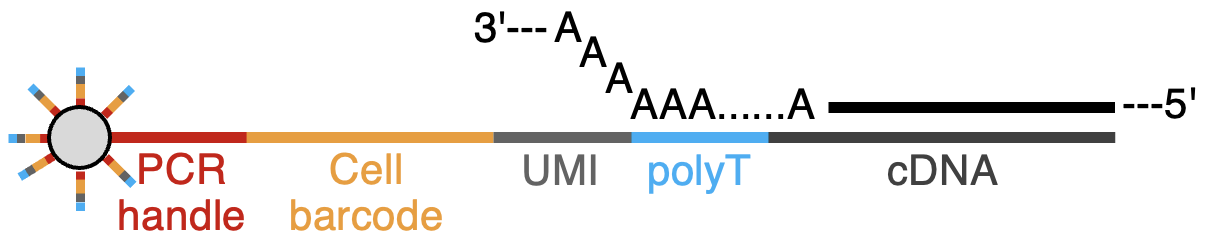
Step 4: cDNA Fragmentation and Adapter Ligation
Full-length cDNAs are fragmented into smaller pieces, and sequencing adapters are ligated to both ends. Importantly, only fragments containing the 5' portion of the original primer (Cell Barcode + UMI etc.) are retained after selection steps, which leads to 3' bias.
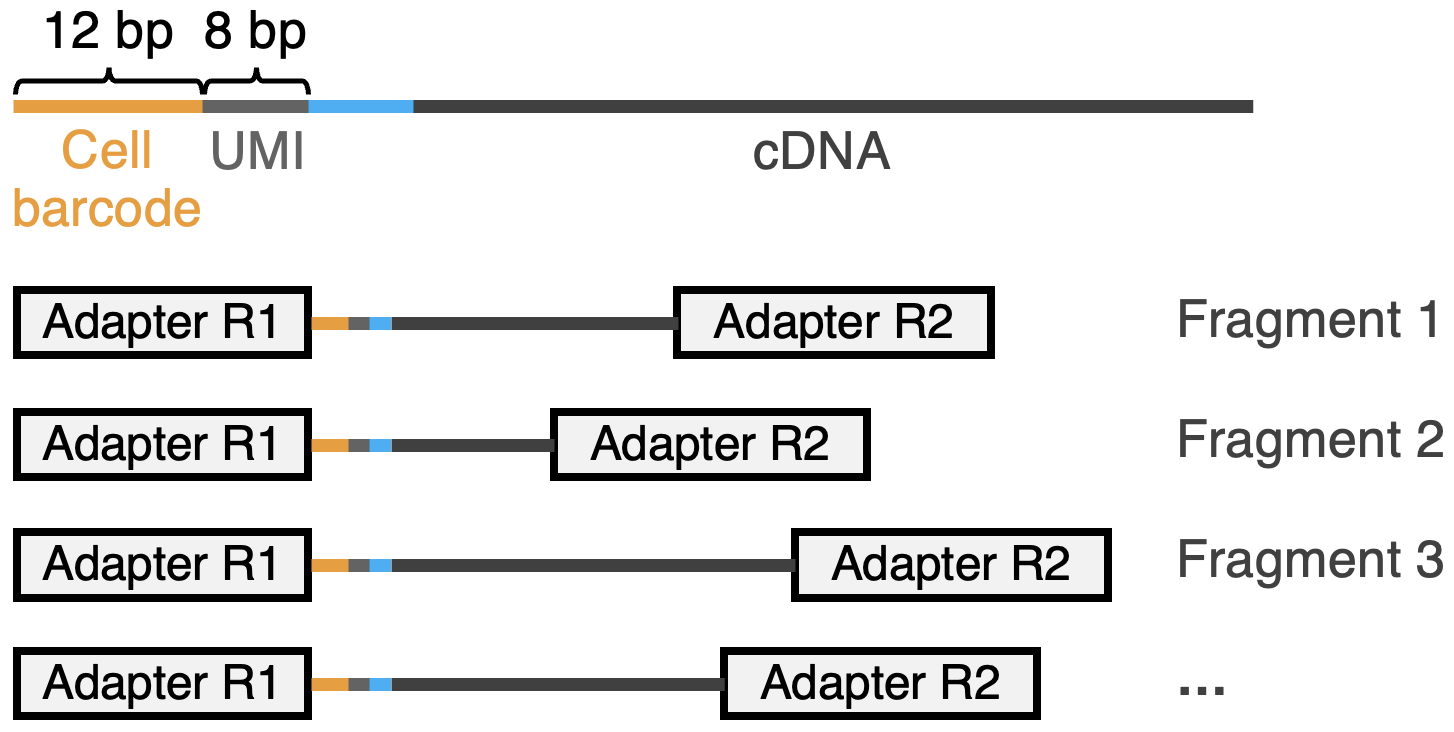
Step 5: Bridge PCR and Library Preparation
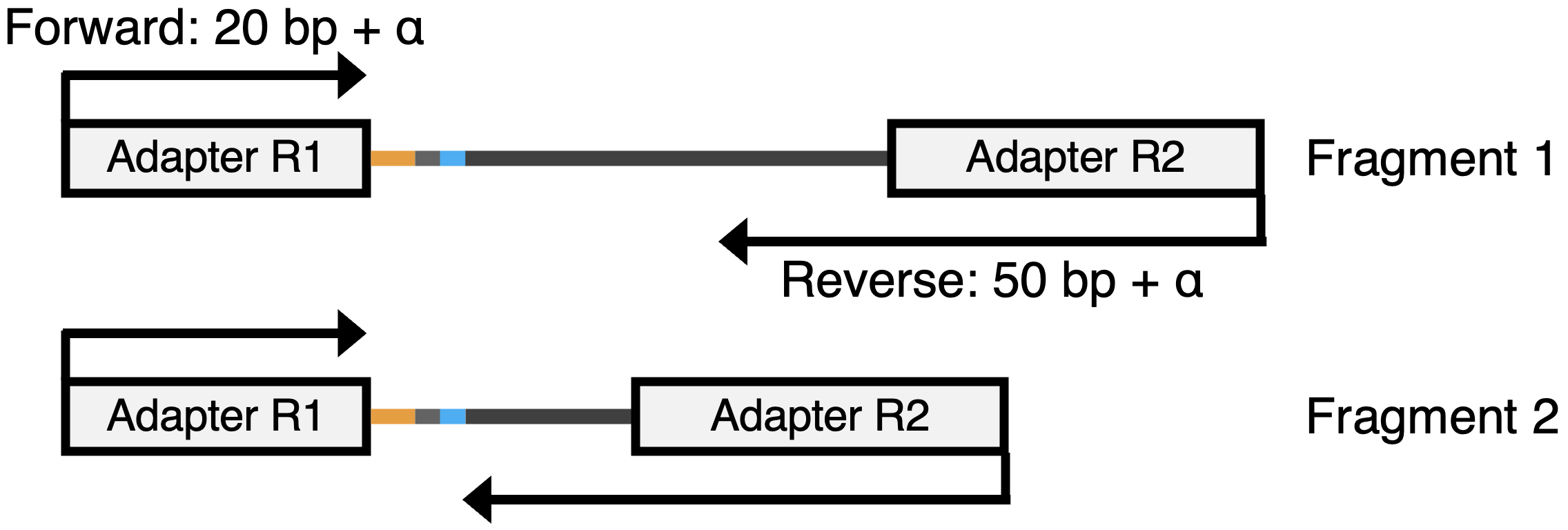
The adapter-ligated cDNA fragments are amplified via bridge PCR, enabling paired-end sequencing (cf. YouTube: Next Generation Sequencing). In this context:
- Forward read R1: Contains Cell Barcode, UMI, and optionally polyT.
- Reverse read R2: Captures the transcript sequence and may include polyA.
Note: Illumina sequencers always read from 5' to 3', so forward reads start at the barcode end, while reverse reads may reach the polyA tail depending on fragment length. In the following example, the reverse read from fragment 2 may reache the polyA. Do you know the reason why?
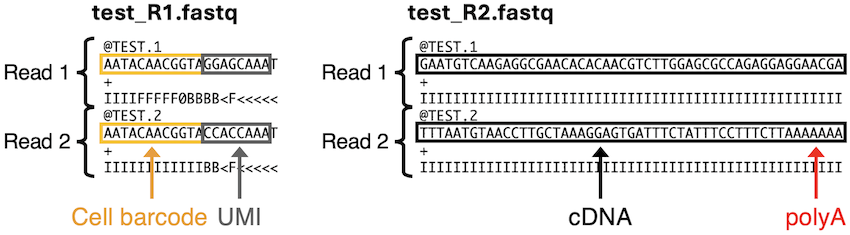
Step 6: Paired-End Sequencing Output
Now you have better understanding for the FASTQ files.
-
test_R1.fastq: Forward reads (Cell Barcode + UMI [+ polyT]) -
test_R2.fastq: Reverse reads (transcript-derived cDNA [+ polyA])